Partager:



Symptômes du syndrome de Smith-Lemli-Opitz, causes et traitement
Le Syndrome de Smith-Lemli-Opitz (SLO) est un trouble métabolique qui comprend plusieurs symptômes différents, tels qu'une croissance significativement lente, des traits caractéristiques caractéristiques, une microcéphalie (taille de la tête inférieure à la normale), un retard mental léger ou modéré, des difficultés d'apprentissage et des problèmes de comportement
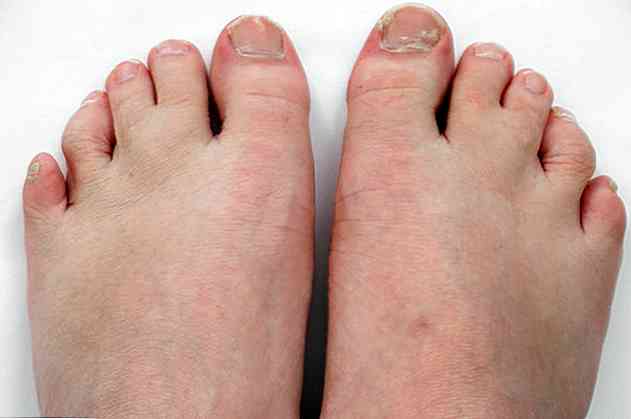
Elle s'accompagne également de malformations pulmonaires, cardiaques, rénales, intestinales et même génitales. En outre, ils peuvent présenter une syndactylie ou une fusion de certains doigts ou une polydactylie; ce qui signifie qu'ils ont plus de 5 doigts sur un pied ou une main.
Cela semble être dû au manque d'enzyme qui est important pour métaboliser le cholestérol qui est acquis par héritage génétique d'un modèle autosomique récessif.
Cependant, ces présentations semblent varier considérablement en fonction de la gravité de la maladie, même dans la même famille.
Ce syndrome peut apparaître dans la littérature sous des noms tels que: déficit en 7-déhydrocholestérol réductase, syndrome RSH ou syndrome SLO.
Un peu d'histoire…
En 1964, les pédiatres David Smith, Luc Lemli et Opitz John ont décrit 3 patients masculins atteints de microcéphalie et d'hypogénitalisme, et ont défini cette condition comme une RSH par les initiales des noms de famille originaux de ces patients.
Par la suite, le nom du syndrome a été changé pour les noms de famille des découvreurs (SLO).
Environ 30 ans plus tard, Tint et al. (1994) ont découvert chez 5 patients atteints de cette affection, des concentrations de cholestérol sanguin significativement faibles, mais une augmentation de plus de 1000 fois les taux de 7-déshydrocholestérol. Ils ont vu que cette augmentation était due à l'absence d'une enzyme qui devrait transformer le 7-déhydrocholestérol en cholestérol.
Plus tard, le gène DHCR7 associé à cette maladie a été identifié, le clonage en 1998 (Witsch-Baumgartner & Lanthaler, 2015).
Statistiques
Le syndrome de Smith-Lemli-Opitz affecte environ 1 à 20 000 à 60 000 naissances vivantes dans le monde. En réalité, il peut être hérité chez une personne sur 1590 à 13 500, mais ce chiffre n'est pas utilisé car de nombreux fœtus atteints de cette maladie meurent avant leur naissance (Organisation nationale des troubles rares, 2016).
En ce qui concerne le sexe, il affecte également les hommes et les femmes, même s'il est plus facile de le diagnostiquer chez les hommes, car les malformations génitales sont plus visibles que chez les femmes. En outre, il semble qu’il soit plus fréquent chez les personnes d’ascendance européenne; en particulier des pays appartenant à l'Europe centrale tels que la République tchèque ou la Slovaquie. Cependant, il est très rare dans la population africaine ou asiatique.
Causes du syndrome de Smith-Lemli-Opitz
Le syndrome SLO apparaît dû à des mutations du gène DHCR7, présent sur le chromosome 11, responsable de l'envoi des ordres de fabrication de l'enzyme 7-déhydrocholestérol réductase. C'est l'enzyme qui module la production de cholestérol et qui serait absente ou très peu présente dans ce syndrome, ce qui conduit à une production insuffisante de cholestérol qui empêcherait une croissance normale.
Cela produit un impact important car le cholestérol est important dans le corps. Il s'agit d'un lipide similaire à la graisse, obtenu principalement par des aliments d'origine animale, tels que les jaunes d'œufs, les produits laitiers, la viande, la volaille et le poisson.
Il est essentiel que l'embryon se développe sans difficulté, en exerçant des fonctions importantes telles que la contribution à la structure des membranes des cellules et de la myéline (substance qui recouvre les cellules du cerveau). Il sert également à produire des hormones et des acides digestifs.
L'absence de l'enzyme 7-déhydrocholestérol réductase provoque l'accumulation dans l'organisme de composants pouvant être toxiques pour le cholestérol. Nous avons donc, d’une part, un faible taux de cholestérol et, en même temps, une accumulation de substances qui peuvent être toxiques pour l’organisme; provoquant un manque de croissance, un retard mental, des malformations physiques et des problèmes dans les organes internes.
Cependant, on ne sait pas avec certitude comment ces problèmes associés au cholestérol donnent lieu à la symptomatologie du syndrome de Smith-Lemli-Opitz.
Actuellement, plus de 130 mutations liées au syndrome ont été trouvées dans le gène DHCR7. En fait, il existe une base de données qui inclut tous les cas décrits de SLO avec ses variantes, ses phénotypes et ses génotypes.
Bien qu'il y ait tant de mutations possibles, la majorité des cas appartiennent aux 5 plus fréquents et les autres sont très rares (Witsch-Baumgartner & Lanthaler, 2015).
Ces mutations dans le gène DHCR7 sont héritées d'un schéma autosomique récessif, ce qui signifie qu'une personne présentant le syndrome doit avoir hérité du gène muté par les deux parents. Si vous ne le recevez que de l'un des parents, vous ne présenterez pas la maladie; mais cela pourrait être un transporteur et le transmettre dans le futur.
Il y a un risque de 25% que les deux parents soient porteurs d'un enfant affecté, alors que le risque que l'enfant soit porteur serait également de 50% à chaque grossesse.Par contre, dans 25% des cas, il peut être né sans ces mutations génétiques ou être porteur; toutes ces données sont indépendantes du sexe du bébé (Organisation nationale des troubles rares, 2016).
Il convient de garder à l'esprit qu'il ya plus de chances d'avoir des enfants atteints d'un trouble génétique récessif si les parents qui sont proches parents (ou de sang) pour les parents qui ne sont pas ces liens.
Quels symptômes avez vous?
Les symptômes varient en fonction de la personne concernée, en fonction de la quantité de cholestérol qu'ils peuvent produire.
Selon Jiménez Ramírez et al. (2001), les caractéristiques cliniques couvrent plusieurs aspects et peuvent être très diverses. Généralement, ils se trouvent sur le visage, les membres et les organes génitaux; bien qu'ils puissent impliquer d'autres systèmes corporels.
Beaucoup de personnes atteintes présentent des caractéristiques typiques de l'autisme, affectant l'interaction sociale. Si la condition est légère, seuls certains problèmes d'apprentissage et de comportement peuvent être observés. Mais dans les cas les plus graves, la personne peut avoir une grande déficience intellectuelle et des anomalies physiques pouvant entraîner la mort.
Il y a des symptômes qui peuvent déjà être présents dès la naissance de l'individu, bien que nous incluions ceux qui se produisent à tous les stades de la vie:
Chez plus de 50% des patients:
- Manque de développement physique observé après la naissance.
- Retard mental (100%).
- Microcéphalie (90%).
- Syndactylie ou fusion de 2 ou 3 orteils (<95%).
- ptose palpébrale, c'est-à-dire que l'une des paupières supérieures est tombée (70%).
- méat situé dans un endroit différent chez les hommes normaux, tels que dans le bas du gland ou d'une connexion entre le tronc scrotum et le pénis. Il est présent dans 70% des cas.
- Fente palatine qui se manifeste par une sorte de trou allongé dans le palais (50%).
- Mâchoire ou micrognathie très réduite.
- Très petit langage (microglossie).
- Oreilles de faible implantation.
- nez court
- Descente incomplète d'un ou des deux testicules.
- Hypotonie ou tonus musculaire faible.
- Troubles de l'alimentation.
- Troubles du comportement: comportements antisociaux, autodestructeurs et violents. Les comportements d'autostimulation autistique apparaissent également comme des mouvements d'équilibrage répétitifs.
- autisme
De 10 à 50% des cas:
- Les premières cataractes.
- Polydactylie ou encore un doigt après le petit doigt.
- Retard de croissance au stade foetal.
- Les organes génitaux ambigus.
- des anomalies cardiaques.
- rein multicystique.
- Absence de rein ou les deux à la naissance.
- maladies du foie.
- hyperplasie surrénalienne
- anomalies pulmonaires.
- transpiration excessive
- Anomalies cérébrales dans les structures situées sur la ligne médiane, telles que développement incomplet du corps calleux, du septum et du vermis cérébelleux.
- Acrocyanose: vasoconstriction cutanée provoquant une couleur bleuâtre dans les mains et les pieds.
- Pieds Equinovar.
- Sténose du pylore (15%)
- la maladie de Hirschprung, qui provoque un manque de motilité intestinale (15%)
- la photosensibilité.
Autres symptômes:
- Obésité ou coma.
- Accumulation de liquide dans le corps du fœtus.
- Altérations dans le développement neurologique.
- Problèmes neuropsychiatriques, qui apparaissent plus fréquemment lorsqu'ils atteignent l'âge adulte.
- Insuffisance respiratoire due à des problèmes pulmonaires.
- perte auditive.
- Altérations de la vision, qui peuvent être accompagnées de strabisme.
- Vomissements
- la constipation
- convulsions.
Comment peut-il être diagnostiqué?
Ce syndrome apparaît dès la conception même si le bébé naît, les symptômes ne sont pas très clairs et sont plus subtils que dans l'enfance ou à l'âge adulte; surtout si elles sont des formes plus douces de la maladie. Pour cette raison, il est détecté tardivement plusieurs fois.
, Est le plus courant mais cette condition et dont on soupçonne peu après la naissance par des malformations habituellement présents (Steiner, 2015).
Selon l'Organisation nationale des troubles rares (2016), le diagnostic repose sur des examens physiques et un test sanguin qui détecte les taux de cholestérol. Il est essentiel que l'enfant soit évalué dans tous les aspects possibles associés à la maladie tels que les yeux, les oreilles, le cœur, les muscles squelettiques, les troubles gastro-intestinaux et des organes génitaux.
En ce qui concerne les analyses de sang, d'un sujet avec SLO aura une forte concentration de 7-déhydrocholestérol (7-DHC) le sang (précurseur à être transformé par l'enzyme 7-déhydrocholestérol réductase pour le cholestérol), et ainsi des niveaux Faible cholestérol
Il peut également être détectée avant la naissance par la technique à ultrasons ou par ultrasons, un dispositif qui utilise des ondes sonores pour examiner l'intérieur de l'utérus enceinte. Avec cette technique, les déformations physiques de ce syndrome peuvent être observées.
L'amniocentèse est un autre test qui consiste à extraire un petit échantillon de liquide amniotique (où le fœtus se développe) pour détecter les anomalies génétiques.La même information peut être obtenue par prélèvement de villosités choriales (CVS) en extrayant un échantillon de tissu du placenta.
En outre, les tests génétiques moléculaires pour le diagnostic prénatal afin de voir s'il y a des mutations dans le gène DHCR7 peuvent être utilisés, et si pour présenter la maladie ou ne sera porteuse.
Quel est le cours de la maladie?
Malheureusement, la plupart des cas les plus graves de SLO meurent peu après la naissance. En cas de déficience intellectuelle grave, il est difficile pour ces personnes de développer une vie indépendante.
Cependant, si des soins médicaux appropriés et une bonne alimentation sont reçus, ces patients peuvent mener une vie normale.
Quels sont les traitements?
Actuellement, il n'y a pas de traitement spécifique pour le syndrome de Smith-Lemli-Opitz. En effet, pas d'origine biochimique de la maladie est connue aujourd'hui pour aujourd'hui avec une certitude absolue puisque le cholestérol a plusieurs fonctions complexes dans le métabolisme.
Le traitement médical du SLO repose sur les problèmes spécifiques rencontrés chez l'enfant affecté et il est préférable d'intervenir rapidement.
Il peut être utile de recevoir des suppléments de cholestérol ou d'augmenter l'apport de celui-ci par l'alimentation, afin d'améliorer le niveau de développement et de réduire la photosensibilité. Parfois, il est combiné avec des acides biliaires.
Pour l’intolérance au soleil, il est conseillé à ces patients d’utiliser des écrans solaires, des lunettes de soleil et des vêtements appropriés lorsqu’ils sortent.
Il a été démontré que la fourniture de médicaments tels que la simvastatine peut réduire la gravité de la maladie. Bien que, comme le phénotype clinique se produit au cours d'une absence de cholestérol dans l'embryogenèse, il doit être administré à ce moment-là (et Lanthaler Witsch-Baumgartner, 2015).
D'autre part, il peut également être utilisé un médicament antagoniste précurseur toxique du cholestérol dans l'excès (7-déhydrocholestérol) pour éviter d'augmenter. Les suppléments de vitamine E peuvent aider.
D'autres types de médicaments spécifiques peuvent être utiles pour des symptômes tels que les vomissements, le reflux gastro-oesophagien ou la constipation.
La chirurgie ou les accolades peuvent être nécessaires si des déformations physiques ou des problèmes musculaires associés à ce syndrome comme une fente palatine, malformations cardiaques, hypotonie musculaire ou des anomalies génitales se produisent.
En conclusion, il est nécessaire de poursuivre les recherches sur ce syndrome afin de développer des traitements plus efficaces et spécifiques.
Références
- Jiménez Ramírez, A. Valdivia Alfaro, R. Hernández González, L. León Corrales, L. Machín Valero, Y. et Torrecilla, L. (2001). Syndrome de Smith Lemli Opitz. Présentation d'un cas avec diagnostic biochimique. Gazette Espirituana, 3 (3).
- Syndrome de Smith Lemli Opitz. (s.f.) Extrait le 6 juillet 2016 de l'Organisation nationale des troubles rares (NORD).
- Syndrome de Smith-Lemli-Opitz. (s.f.) Extrait le 6 juillet 2016 de l'Université de l'Utah, Health Sciences.
- Syndrome de Smith-Lemli-Opitz. (s.f.) Récupéré le 6 juillet 2016 de Counsyl.
- Syndrome de Smith-Lemli-Opitz. (5 juillet 2016). Récupéré de Genetics Home Reference.
- Steiner, R. (1er avril 2015). Syndrome de Smith-Lemli-Opitz. Obtenu à partir de Medscape.
- Tint, G.S., Irons, M., Elias, E.R., et al. (1994). Biosynthèse du cholestérol défectueuse associée au syndrome de Smith-Lemli-Opitz. N Engl J Med, 330: 107-113
- Witsch-Baumgartner, M. et Lanthaler, B. (2015). Anniversaire d'un syndrome: 50 ans du syndrome de Smith-Lemli-Opitz. European Journal of Human Genetics, 23 (3), 277-278.