Partager:



Symptômes, causes et traitements du syndrome de Pearson
Le Syndrome de Pearson C'est l'une des maladies rares connues en raison de sa faible prévalence. Il s'agit d'une maladie de type mitochondrial qui affecte l'ensemble du corps, c'est-à-dire que son affectation est multi-systémique. Son apparition se produit dans l'enfance et se produit en raison de la suppression de l'ADN mitochondrial.
Ce syndrome a été décrit pour la première fois en 1979 par Howard Pearson, pédiatre spécialisé en hématologie. Une décennie plus tard, les délétions de l'ADN mitochondrial à l'origine de ce syndrome ont été découvertes.
Causes du syndrome de Pearson
Cette maladie multisystémique est causée par une anomalie de la phosphorylation oxydative, qui est le processus métabolique par lequel l'énergie libérée par l'oxydation des nutriments est utilisée pour produire de l'adénosine triphosphate (ATP). L’anomalie de ce processus est due à la duplication de l’ADN mitochondrial.
En dépit d'être une maladie mitochondriale, c'est-à-dire transmise par la mère, il a été conclu que le syndrome de Pearson est généralement sporadique. Par conséquent, il existe des délétions d'ADN mitochondrial qui servent de critères diagnostiques, mais la distribution aléatoire de ce type d'ADN entraîne la convergence des cellules normales et d'autres des mutations.
Ce fait, appelé hétéroplasmie, qui survient lorsqu'un individu présente un mélange de différentes populations de mitochondries, est la cause de la grande variabilité de l'expression clinique de la maladie. Ce terme fait référence au fait que, bien que répondant à un même diagnostic, des individus différents présentent des symptômes différents, ainsi que différents niveaux d'affectation.
Quelle est sa prévalence?
Étant une maladie rare, elle touche une minorité de la population. Selon le portail européen des maladies rares, Orphanet, le syndrome de Pearson a une prévalence inférieure à 1/1 000 000.
En outre, il ajoute qu'il n'y a pas plus de 60 cas décrits. Le type d'héritage transmis par le syndrome de Pearson, car il n'est pas lié au sexe, affecte les garçons et les filles de la même manière.
Quels sont ses symptômes?
L'apparition du syndrome de Pearson est au stade infantile et il existe peu de cas de nouveau-nés. Les premiers signes sont visibles pendant la période de lactation et avant six mois de la vie.
Ce syndrome présente une image très variée, avec des conditions différentes. Trois personnes présentent le syndrome de Pearson et présentent les trois caractéristiques suivantes:
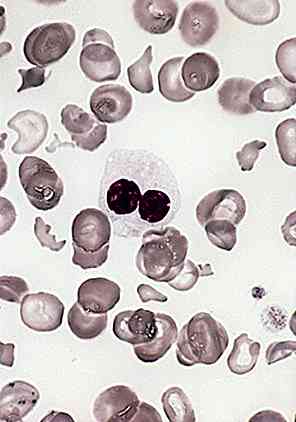
Anémie sidéroblastique réfractaire
C'est le symptôme par excellence du syndrome de Pearson et implique l'altération de la synthèse de l'hémoglobine dans les précurseurs de la moelle osseuse. De cette manière, des sidéroblastes annelés sont produits.
Pour son traitement, il convient de contrôler l'anémie et, en outre, d'éviter la surcharge en fer.
Parfois, cette anémie est associée à une neutropénie profonde qui consiste en la diminution du nombre de neutrophiles (communément appelés leucocytes ou globules blancs).
Également de thrombocytopénie; lorsqu'il existe une situation hématologique anormale et que le nombre de plaquettes est inférieur. Il se produit en raison de la destruction du tissu érythrocytaire dans la moelle osseuse.
Vacuolisation des précurseurs de la moelle osseuse
Les cellules précurseurs de la moelle osseuse, dans le cas du syndrome de Pearson, augmentent considérablement leur taille.
Dysfonction exocrine du pancréas
Ce dysfonctionnement est l'incapacité du pancréas exocrine à exécuter normalement les fonctions digestives. Il est généralement produit par une réduction soudaine de la sécrétion pancréatique. Il est étroitement lié à une mauvaise digestion et, par conséquent, conduit à une malabsorption des aliments non digérés qui déclenchent souvent un état de malnutrition.
Il existe une grande variabilité dans l'expression du syndrome de Pearson, du fait que les cellules pathogènes sont associées aux cellules normales. Pour qu'une personne présente des manifestations pathologiques, il doit accumuler une quantité suffisante d'ADN muté. Parfois, en raison des différents organes et systèmes affectés, on pense que le syndrome de Pearson est une association "incohérente" de symptômes.
Dans une publication de l’hôpital universitaire Doce de Octubre de Madrid, qui consistait en une étude de trois cas de syndrome de Pearson, ils révèlent que d’autres affections qui ont tendance à apparaître plus tard sont les affections oculaires, endocriniennes, cardiaques et neurologiques. En ce qui concerne les maladies cardiaques, certains patients ont nécessité l'implantation d'un stimulateur cardiaque.
Dans une moindre mesure, certains patients atteints du syndrome de Pearson présentent des altérations cérébrales et / ou du tronc cérébral visibles par imagerie par résonance magnétique.
En outre, certains d’entre eux présentent l’hiperlactatorraquia, également connu sous le nom d’hypoglucorraquia, qui suppose la diminution des taux de glucose dans le liquide céphalo-rachidien.En outre, l'hyperprotéinorraquie, l'augmentation des protéines dans le liquide céphalo-rachidien et la diminution de l'acide folique dans ce liquide sont fréquents.
Comment diagnostiquer le syndrome de Pearson?
Normalement, le diagnostic peut être établi en fonction des symptômes observés. Toutefois, et comme l’indique l’Association Pearson Syndrome, il est nécessaire d’effectuer différents tests et examens pour conclure le diagnostic de ce syndrome.
En premier lieu, lorsqu'un syndrome mitochondrial est suspecté, une analyse préventive peut être effectuée pour déterminer les altérations génétiques les plus fréquentes dans l'ADN mitochondrial.
Un autre test très important dans le syndrome de Pearson est la biopsie musculaire et dans le cas où différents symptômes se rencontrent, il est essentiel. Ce test implique le retrait d'un petit échantillon de tissu musculaire à examiner et à analyser. C'est un test rapide et peu invasif et ce n'est pas douloureux.
La neuroradiologie est utile dans le diagnostic de ce syndrome car elle offre des images de l'état du cerveau et il sera possible de détecter l'existence d'une anomalie. Merci aux études de laboratoire, les niveaux d'acide lactique et le liquide céphalorachidien peut être mesurée et établir ainsi si les moyens correspondent, s'il y a des niveaux d'anomalie.
Enfin, des tests analysant l’activité des enzymes sont effectués.
Dans les cas où il existe des symptômes cardiaques ou qui affectent d'autres organes ou systèmes, tels que la vision, les tests correspondants seront effectués afin d'appliquer le traitement dont ils ont besoin. Des études gastro-entérologiques et nutritionnelles peuvent également être réalisées pour vérifier que l'absorption des nutriments est effectuée correctement.
Traitement
À ce jour, le syndrome de Pearson nécessite un traitement symptomatique. C'est-à-dire qu'il n'existe pas de thérapie ou de médicament pour guérir la maladie et que, par conséquent, les traitements visent à soulager les symptômes que ce syndrome provoque chez les personnes qui en souffrent.
A cette fin et, d'abord, il est très important d'avoir procédé à une analyse exhaustive fournit des données sur l'état de santé de l'enfant et quelles sont leurs faiblesses de se concentrer sur la façon la plus appropriée de traitement. De plus, des contrôles médicaux sont nécessaires pour vérifier l'évolution et vérifier que le traitement utilisé est approprié.
Normalement, le traitement visera à soulager les épisodes infectieux et les problèmes métaboliques.
Dans les cas où l'anémie est grave, des transfusions sanguines seront prescrites. Dans certains cas, ce traitement sera accompagné par traitement à l'érythropoïétine consistant à appliquer une hormone qui contribue à la création de globules rouges également connu sous le nom érythrocytes.
En outre, si des troubles ou des symptômes endocriniens affectant d'autres organes qui n'ont pas été mentionnés dans cette section et que j'ai mentionnés ci-dessus, tels que le système visuel, le cœur, etc., seront traités.
Est-ce mortel?
Malheureusement, le syndrome de Pearson met habituellement fin à la vie de ces enfants avant l'âge de trois ans. Les causes sont variées et parmi elles:
- Risque de sepsie qui est la réponse massive du corps à un processus infectieux.
- Crises métaboliques avec acidose lactique ou insuffisance hépatocellulaire.
Il n'y a pas de chiffres qui nous renseignent sur le taux de survie des enfants touchés par ce syndrome. Mais, dans le cas où ces mineurs survivent à la symptomatologie, le syndrome de Pearson disparaît en raison de l'évolution phénotypique, faisant disparaître spontanément les symptômes hématologiques.
En ce qui concerne les signes neurologiques et myopathiques, ils peuvent augmenter ou disparaître. Dans certains cas, le syndrome de Pearson entraîne une autre maladie mitochondriale, le syndrome de Kearns-Sayre.
Quel est le syndrome de Kearns-Sayre?
Ce syndrome, également de type mitochondrial, l'ophtalmoplégie externe progressive (faiblesse progressive des muscles oculaires et soulève la paupière), la rétinite pigmentaire (groupe de maladies dégénératives de l'œil) et son début est avant 20 ans est caractérisée. Parmi les autres caractéristiques communes, citons la surdité, l'ataxie cérébelleuse et le blocage cardiaque.
Orphanet estime que le syndrome de Kearns-Sayre touche une personne sur 125 000.
La maladie se produit généralement pendant l'enfance avec des symptômes suivants: ptose (décollement total ou partiel d'un organe), la rétinopathie pigmentaire et ophtalmoplégie externe progressive. Ensuite, d'autres symptômes apparaissent selon la distribution de l'anomalie moléculaire, comme dans le syndrome de Pearson.
D'autres symptômes associés à ce syndrome sont la surdité neurosensorielle bilatérale, Affectations cardiaques, les effets du système nerveux central (ataxie cérébelleuse, dysarthrie, faiblesse faciale bilatérale, déficit intellectuel), la myopathie des muscles squelettiques, les troubles intestinaux et endocriniens (puberté retardée , hypoparathyroïdie, diabète) et insuffisance rénale. La progression de la maladie est lente et peut durer des décennies. Durant ces années, de nouveaux symptômes peuvent apparaître ou aggraver ceux qui sont déjà présents.
Le syndrome de Kearns-Sayre est également causé par des délétions de fragments d’ADN mitochondrial, affectant le processus de phosphorylation oxydative. Il existe des cas exceptionnels de ce syndrome qui surviennent sans la suppression de l'ADN mitochondrial et qui résultent de mutations ponctuelles localisées.
Le diagnostic est généralement établi en fonction des manifestations et, par la suite, des tests effectués pour le confirmer. Les tests sont généralement les mêmes que dans le cas du syndrome de Pearson. Normalement, le diagnostic n'est pas effectué au stade prénatal.
La plupart des cas de ce syndrome surviennent de manière sporadique. Les délétions de l'ADN mitochondrial sont transmises d'une génération à l'autre, exceptionnellement. On estime que moins de 4% des femmes transmettent leur progéniture à une délétion de l'ADN mitochondrial. Dans le cas des hommes, ils ne le transmettent pas.
De même, le traitement de ce syndrome tente d’atténuer les symptômes provoqués. Des examens réguliers effectués par un spécialiste du cœur sont recommandés. Dans les cas où les blocages cardiaques, nécessiteront la mise en œuvre du dispositif de stimulateur cardiaque ou un défibrillateur pour améliorer la qualité de vie de ces patients.
Les patients sourds peuvent utiliser des appareils auditifs. En outre, le supplément de coenzyme Q10 s'est révélé bénéfique dans certains cas. Dans le cas des manifestations ophtalmiques, ils peuvent être traités par chirurgie, bien que le risque de récidive et les complications oculaires potentiels est élevé.
Le pronostic de la personne atteinte du syndrome de Kearns-Sayre dépendra des organes touchés et du degré d’implication dans chacun d’entre eux. Ce fait est étroitement lié à la proportion d'ADN mitochondrial affecté et en bonne santé qui se trouve dans chacun d'eux.
Dans un grand nombre de cas, l'espérance de vie des personnes atteintes de ce syndrome peut être normale si elles reçoivent des soins médicaux adéquats, en suivant le traitement et les directives prescrits par les professionnels de la santé.
Bibliographie
- McShane, M.A. (1991) Syndrome de Pearson et encéphalomyopathie mitochondriale chez un patient présentant une délétion d'ADNmt. Département de neurologie, Hospital for Sick Children, Queen Square, Londres.
- Syndrome de Kearns-Sayre. Orphanet (2014).
- Syndrome de Pearson Orphanet (2006).
- Cánovas, R. de la Prieta, J.J. Alonso, C. Ruiz, T. Pereira, C. Aguirre. Anémies sidéroblastiques (2001). Service et chaire de médecine interne. UPV / EHU. Hôpital Cruces. Barakaldo.
- Martín Hernández, M.T. García Silva, P. Quijada Fraile, A. Martínez de Aragon, A. Cabello, M.Á. Martín Syndromes Pearson et Kearns-Sayre: deux maladies mitochondriales multisystèmes, en raison de suppressions dans l'ADN mitochondrial (2010).
- Cammarata-Scalisi, F., Lopez-Garcia, E., empereur, S., Ruiz-Pesini, E., Da Silva, G., Camacho, N., Montoya, J. Syndrome de Pearson Rapport d'un cas (2011).